When a DNA Repair Gene Turned Rogue: A Cancer Vulnerability
Cancer is often described as a disease of broken brakes — cells that ignore stop signals and accelerate toward chaos. But sometimes the brakes themselves fracture in unexpected ways. One class of genes whose failure both causes tumors and paradoxically makes them vulnerable are DNA repair genes. When one of these genes "goes rogue" — meaning it becomes mutated or inactivated — it can create a catastrophic dependence in the tumor that clinicians can exploit. This article follows how scientists turned that molecular misstep into a treatment strategy, the biology behind it, and what it means for the next decade of cancer care.
Cancer cell DNA damage
DNA Repair: The Cell’s Error-Correction Machinery
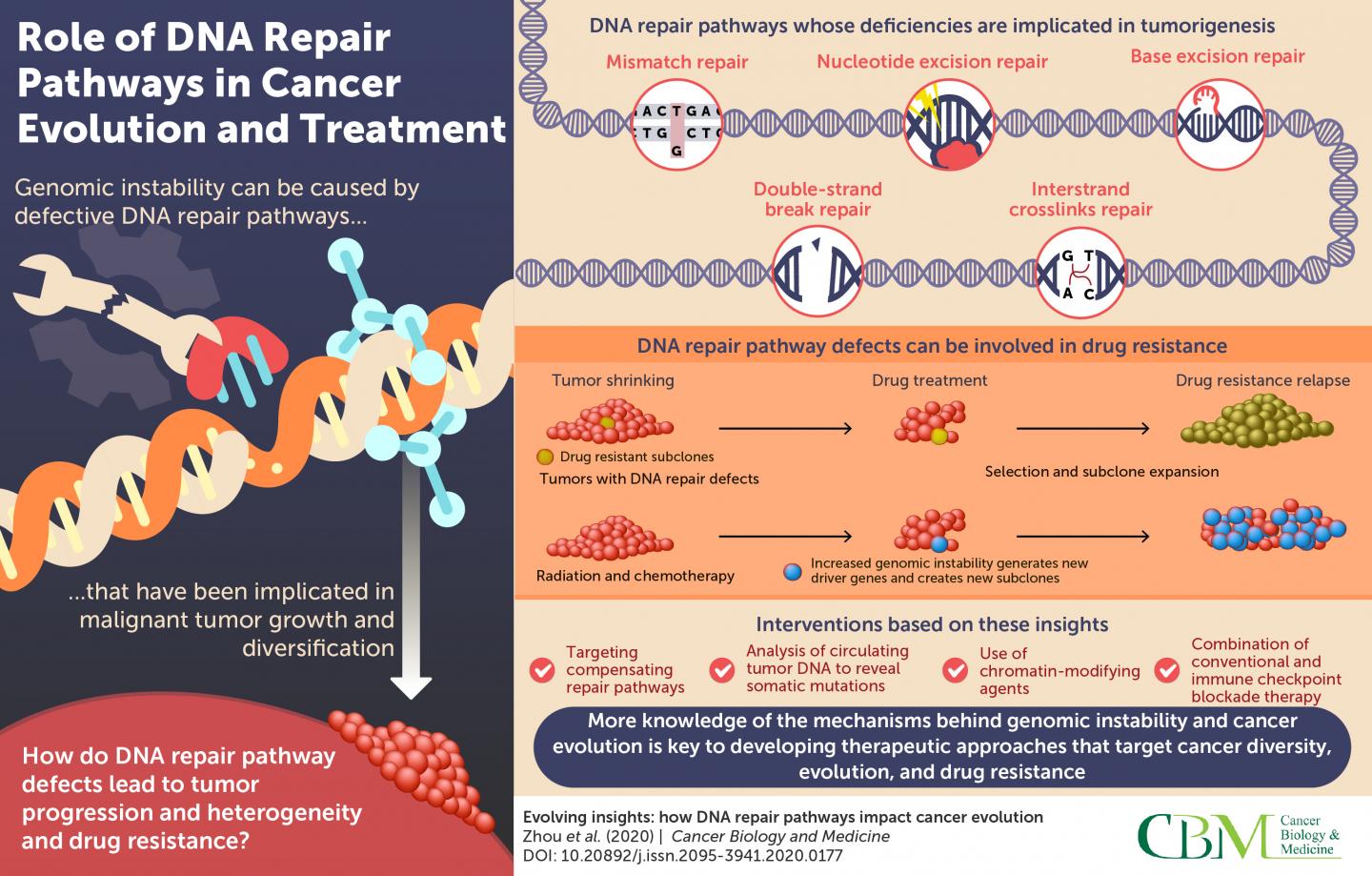
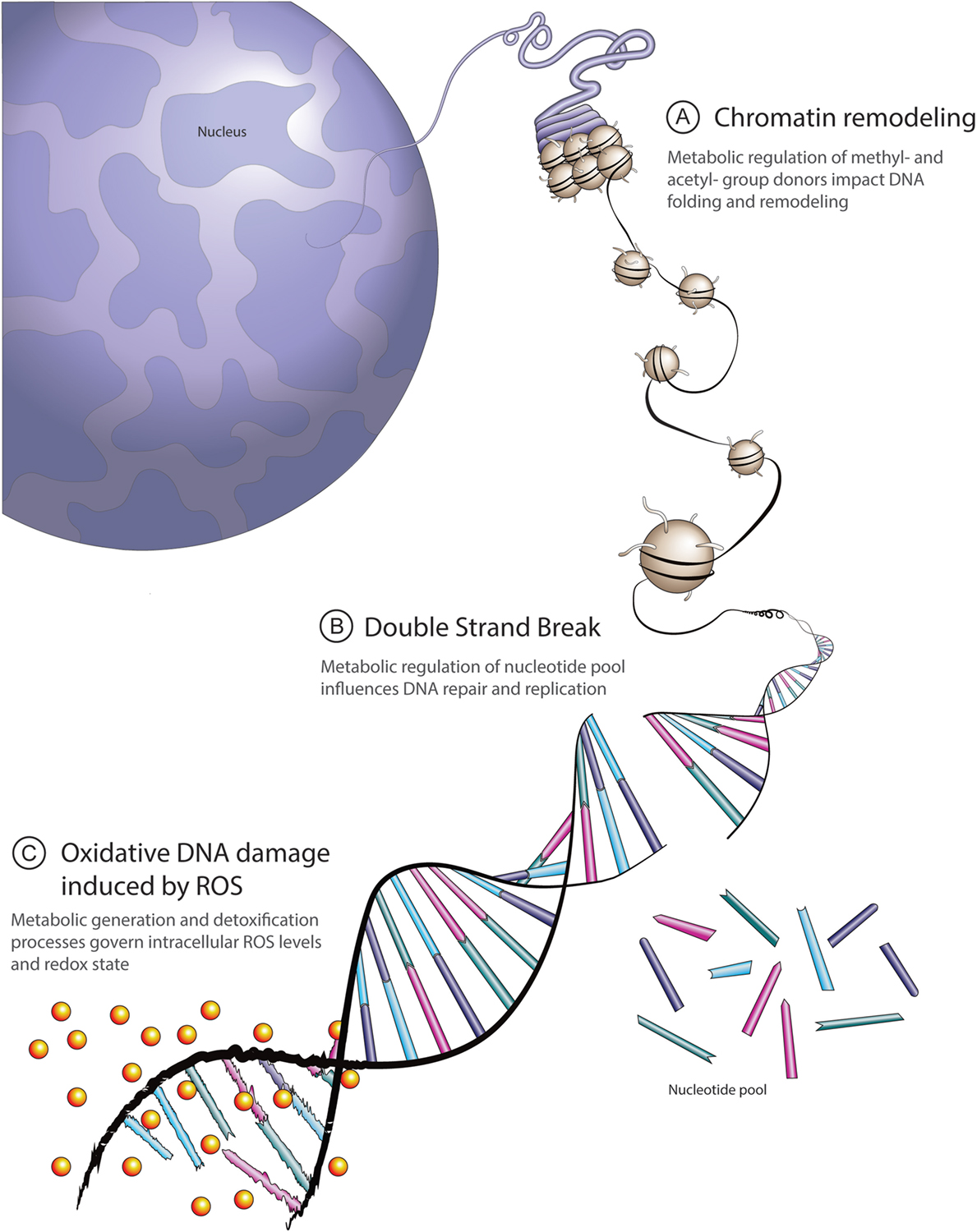
Every time a cell divides or reads a gene it risks copying errors: broken strands, misplaced bases, gaps. To manage this, our cells deploy a network of DNA repair pathways that recognize and fix different kinds of damage. Think of them as specialized repair crews: some patch single-base errors, others stitch together broken double strands. Together they preserve genomic integrity, prevent mutations from accumulating, and keep cells functioning properly.
DNA repair pathways diagram
The Usual Suspects: Major Repair Pathways Explained
When a repair gene is faithful, the genome stays stable. When key players fail, mutations rise, and cancer can follow. The most relevant pathways for this story are:
Homologous recombination repair
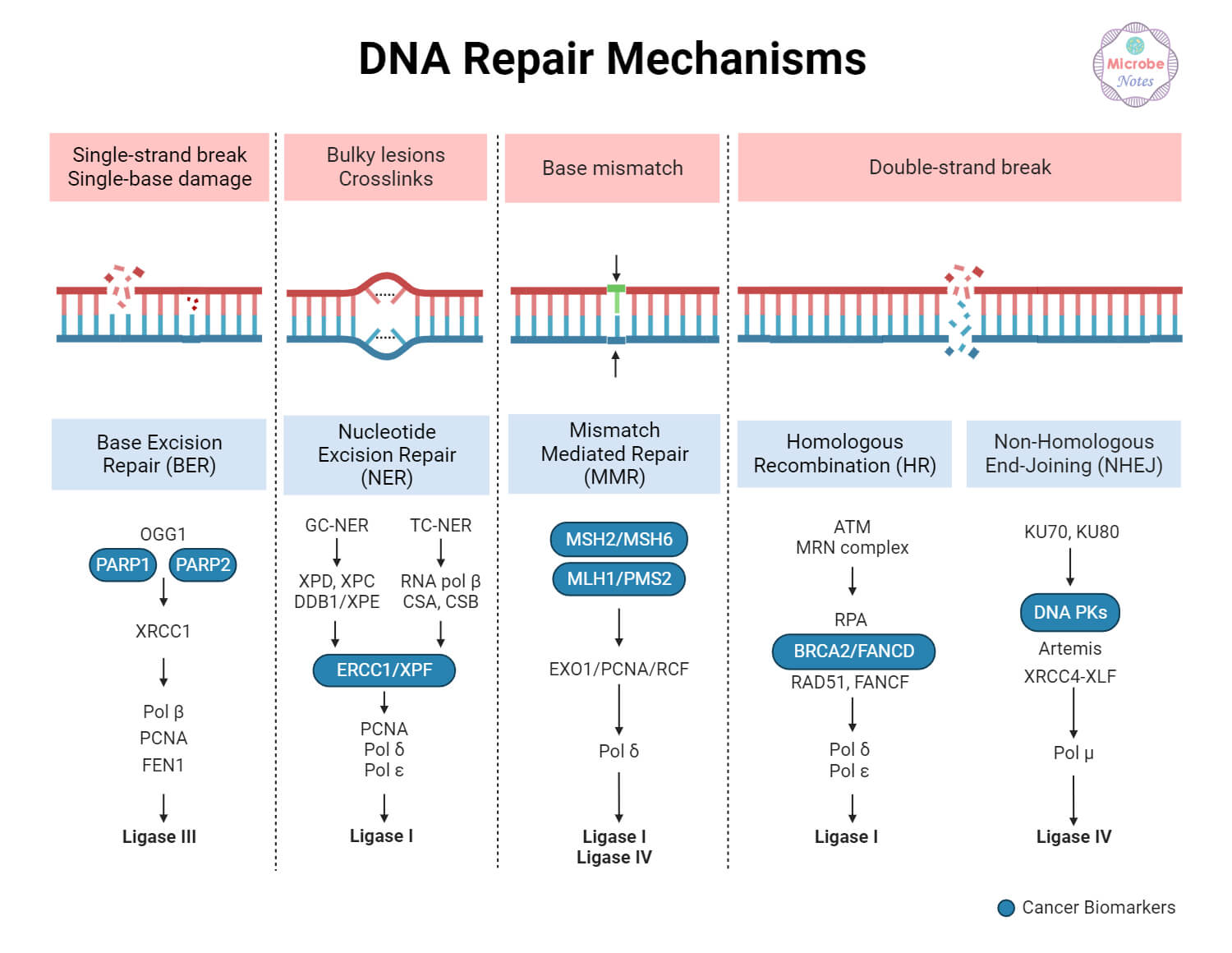
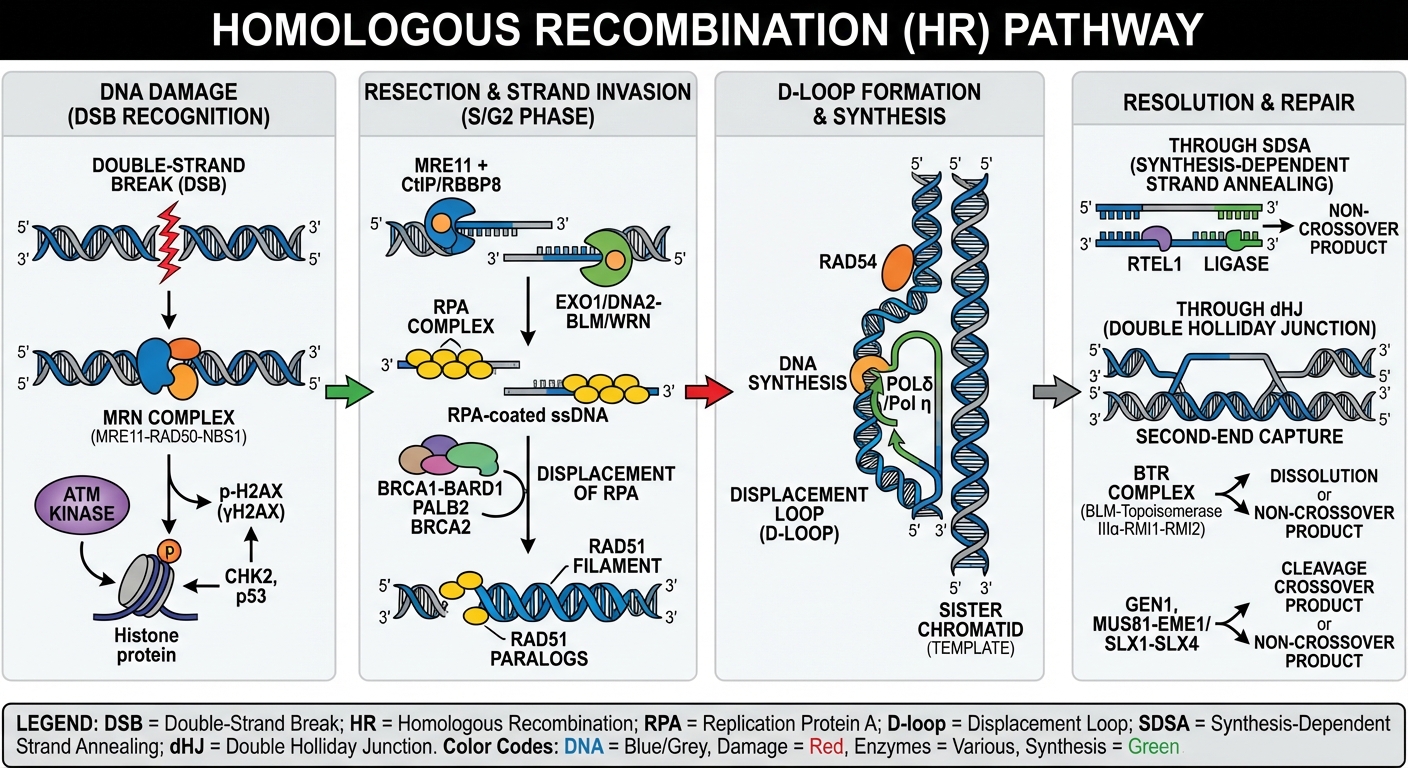
- Homologous recombination (HR): a high-fidelity repair route for double-strand breaks that requires a sister chromatid as a template. Genes like BRCA1, BRCA2, RAD51, and PALB2 are core components.
- Non-homologous end joining (NHEJ): a faster but error-prone double-strand break repair system that rejoins broken ends without a template.
- Base excision repair (BER) and single-strand break repair: processes that correct small chemical changes or single-strand disruptions. PARP enzymes are central to sensing and coordinating repair here.
- Mismatch repair (MMR): corrects mispaired bases that slip in during DNA replication; defects here lead to high mutation rates and microsatellite instability.
Mutations in any of these pathways can seed cancer — but they also create predictable weaknesses.
How a 'Rogue' Gene Revealed a Therapeutic Blind Spot
The classic example of a repair gene gone wrong is BRCA1 or BRCA2. In their normal state, these genes help faithfully repair double-strand breaks through homologous recombination. When they are mutated — whether inherited in the germline or lost through somatic change in tumor cells — HR function collapses. Cells compensate imperfectly, accumulate mutations, and can become malignant.
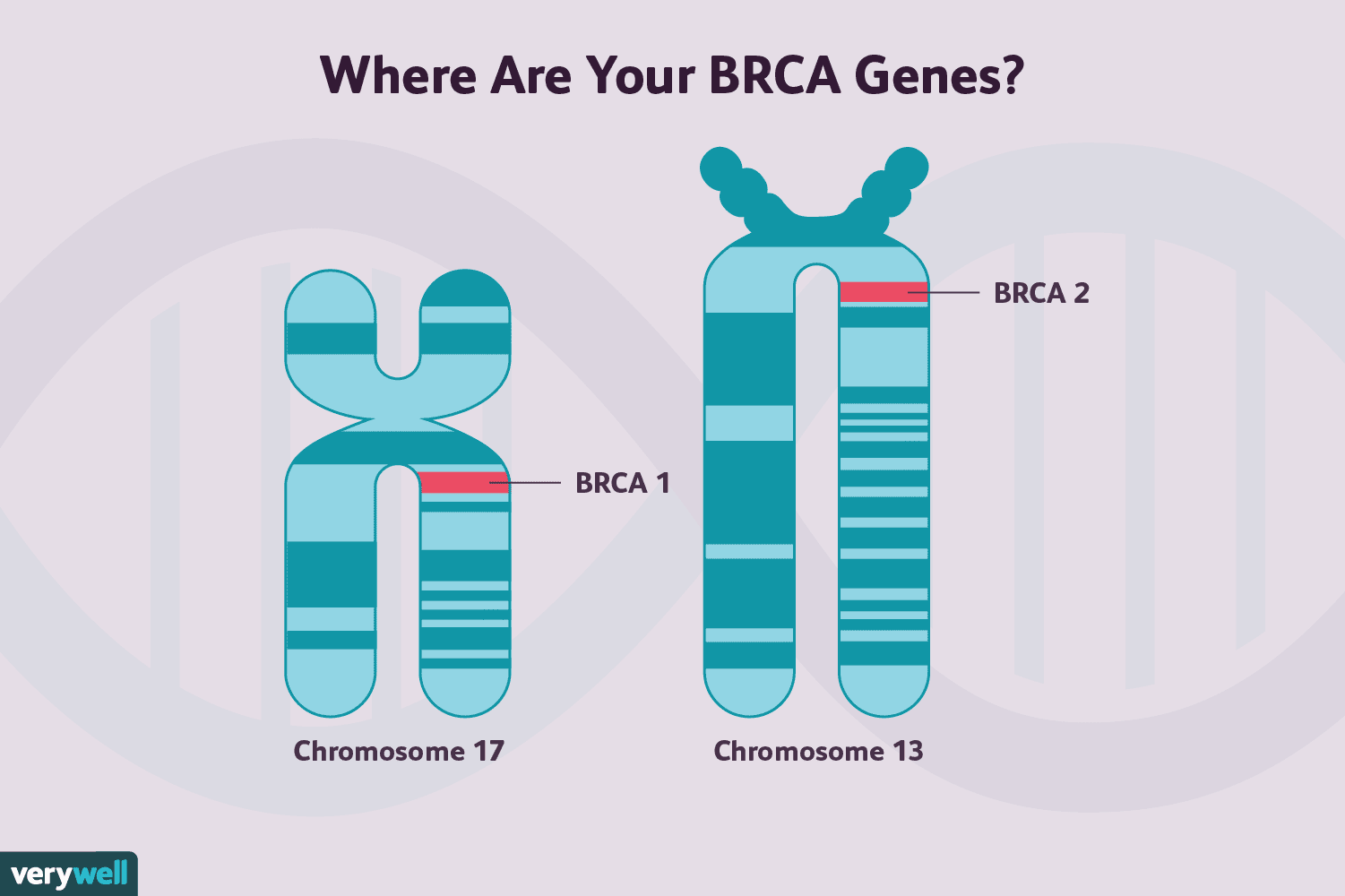
BRCA gene mutation illustration
When the high-fidelity repair line collapses, cancer cells often lean on lower-fidelity systems; that reliance becomes their Achilles' heel.
Researchers realized that tumors with HR deficiency (HRD) become hyperdependent on other repair mechanisms to survive. One of those backup systems relies on PARP enzymes to fix single-strand breaks. Block PARP in an HR-deficient tumor and DNA damage accumulates until the cell can no longer cope — triggering catastrophic cell death. This is the concept of synthetic lethality: two simultaneous defects kill a cell, while either defect alone is survivable.
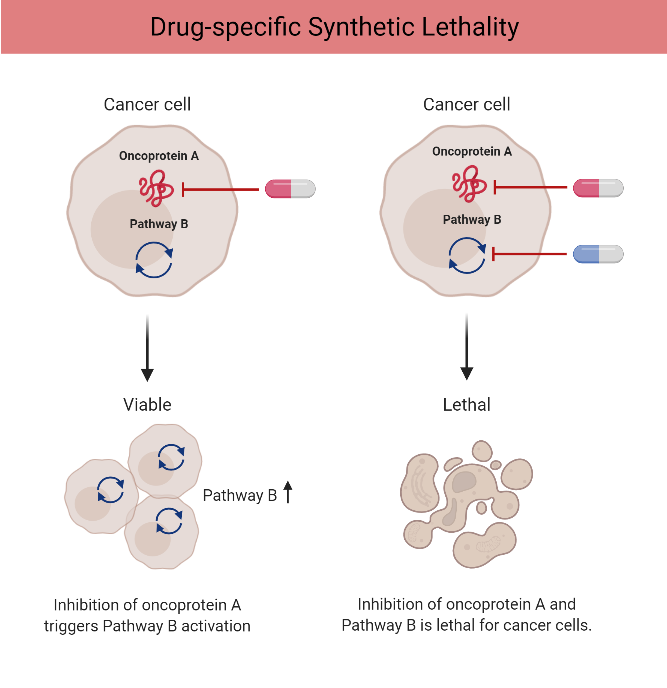
Synthetic lethality mechanism
From Mechanism to Medicine: Exploiting Synthetic Lethality
Translating synthetic lethality into a therapy required three things: a drug that targeted the compensatory pathway, a clear idea of which tumors depended on that pathway, and clinical trials demonstrating benefit. PARP inhibitors — small molecules that block PARP activity and trap PARP on DNA — fit the first requirement. The second requirement came from genetic testing: tumors with BRCA mutations or other HRD markers were the expected responders.
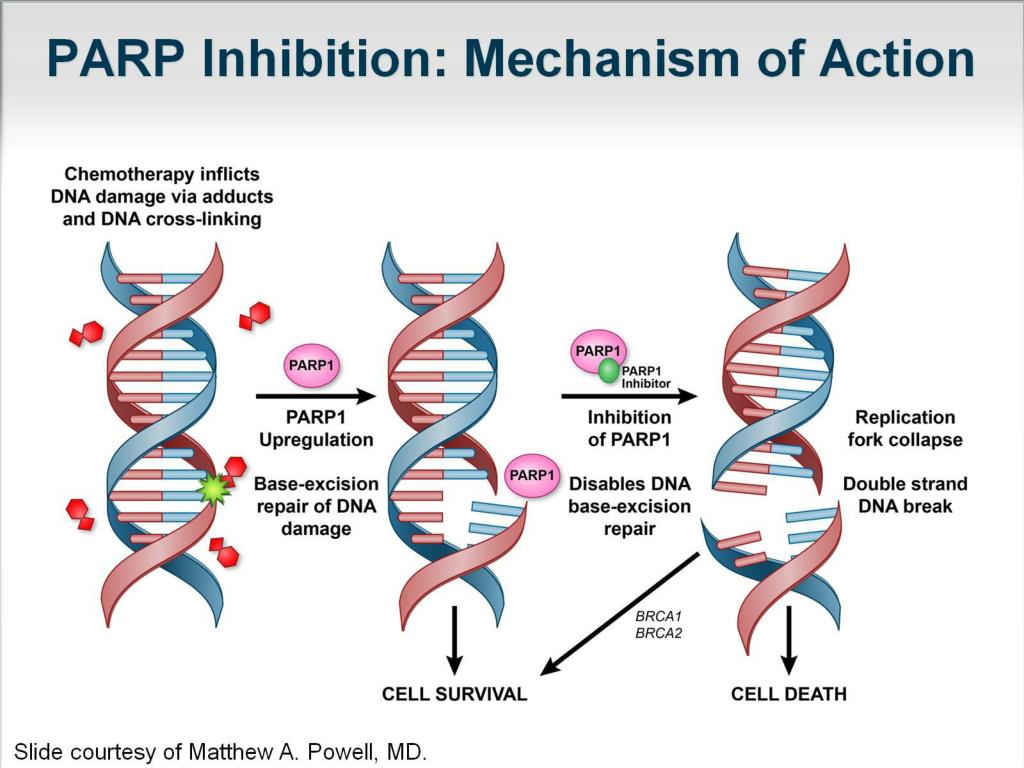
PARP inhibitors cancer therapy
The result has been a paradigm shift: rather than targeting the cancer’s growth signals directly, clinicians target a vulnerability created by the tumor’s genetic weakness. For many patients with BRCA-mutant ovarian, breast, pancreatic, or prostate cancers, PARP inhibitors produced meaningful responses and durable disease control where conventional therapies fell short.
Beyond BRCA: A Wider Landscape of Vulnerabilities
BRCA mutations were the proof of principle, but they are not the only way tumors can be HR-deficient. Loss of PALB2, RAD51 paralogs, or functional inactivation of ATM or ATR can produce an HRD-like state. Laboratories now use composite HRD scores — genomic scars that reflect past repair failures — and panel sequencing to identify tumors that may respond to the same synthetic-lethal approach.
Expanding the net has consequences: more patients become eligible for targeted therapy, but identifying true responders is complex. Not every tumor with a listed mutation behaves the same; context, co-mutations, and tumor evolution matter.
A Table to Clarify Repair Roles
To understand how different genes fit into repair and therapy decisions, consider a concise comparison:
| Pathway | Key Genes | Therapeutic Implication |
|---|---|---|
| Homologous recombination | BRCA1, BRCA2, PALB2, RAD51 | HRD predicts PARP inhibitor sensitivity |
| Base excision repair | PARP1/2 | PARP inhibitors block compensation for HRD |
| Mismatch repair | MLH1, MSH2, MSH6 | MMR-deficient tumors have high mutation burden; immunotherapy relevance |
Clinical Translation: Testing, Treatment, and Patient Selection
Modern oncology now routinely integrates genomic testing. Patients with advanced cancers are offered multigene panels or tumor sequencing to find actionable alterations. A detected BRCA mutation — whether inherited or somatic — can steer a patient toward a PARP inhibitor at the appropriate disease stage. Likewise, HRD scores, loss-of-function signatures, and functional assays are moving into practice to broaden who may benefit.
Genomic testing cancer diagnosis
That said, selection is not binary. Clinicians weigh prior treatments, tumor type, and performance status. Combining PARP inhibitors with chemotherapy or radiation can increase effectiveness because those modalities generate DNA damage that the weakened tumor struggles to repair. But combinations also increase toxicity, and balancing efficacy against side effects remains a central clinical challenge.
Resistance: When the Weakness Disappears
Tumors are adaptable ecosystems. Under the selective pressure of therapy, some cancer cells acquire secondary changes that restore enough repair function to survive. For example, a tumor may develop a reversion mutation that restores the BRCA open reading frame, re-enabling homologous recombination. Other mechanisms include increased drug efflux, stabilization of alternative repair proteins, or rewiring of replication stress responses.
Resistance forces clinicians and researchers to iterate: combine agents to prevent escape, develop next-generation inhibitors that overcome common resistance mechanisms, and refine biomarkers that predict when resistance is emerging rather than established.
Expanding the Arsenal: New Targets and Combinations
The success story of PARP inhibitors inspired a broader hunt for synthetic-lethal partnerships. Drugs targeting ATR, CHK1, DNA-PK, and WRN aim at vulnerabilities created by replication stress or specific repair defects. Immune checkpoint inhibitors intersect with repair deficiencies too: tumors with high mutation loads from repair failure may be more visible to the immune system and thus better immunotherapy candidates.
Combination strategies now in development include:
- PARP inhibitors plus immune checkpoint blockade to exploit mutation-driven neoantigens.
- PARP inhibitors with ATR or CHK1 inhibitors to overwhelm residual repair and replication stress responses.
- Targeted agents with epigenetic drugs that deepen repair defects or silence compensatory pathways.
Each approach raises new questions about toxicity, scheduling, and which patients will tolerate multi-agent regimens.
The Role of Functional Assays and Real-Time Monitoring
Genomic sequencing gives a static picture — a list of past or present mutations. Functional assays that measure repair capacity in living cells, circulating tumor DNA that reports emerging resistance, and imaging biomarkers of DNA damage are powerful complements. Their adoption could allow clinicians to switch strategies before relapse becomes clinically apparent.
Ethical and Practical Considerations
Testing for repair gene defects has ripple effects beyond treatment choices. Finding a germline mutation implicates family members and raises questions about screening, preventive surgery, and insurance. Access to testing and targeted drugs remains uneven globally, and cost considerations affect which patients receive these therapies. Equity of access is a medical and moral priority as precision oncology advances.
What Patients Should Know
For patients and families, the headline is hopeful: a mutation that helped a tumor grow may also be the reason it responds to a precision drug. But the pathway is not guaranteed. Key points for patients to discuss with clinicians include:
- Comprehensive testing: Ask whether both tumor and germline testing have been offered.
- Trial opportunities: If approved options are limited, clinical trials may provide access to new combinations or next-generation drugs.
- Monitoring plan: Understand how response and resistance will be tracked and what the next steps might be.
Future Directions: From Vulnerability to Durable Control
Scientists are working on several fronts to make vulnerability-targeting more durable. Better biomarkers will predict who benefits most and when to change strategy. More intelligent drug sequencing and adaptive therapy approaches aim to suppress resistant clones rather than simply eradicating the bulk tumor. Advances in single-cell sequencing, liquid biopsies, and systems biology will reveal how tumors rewire themselves and where new vulnerabilities emerge.
Moreover, the synthetic-lethality framework is expanding beyond DNA repair. Researchers search for context-specific dependencies across metabolic pathways, epigenetic regulators, and stress-response networks. Each new pair of complementary defects is a potential route to selective therapy that spares healthy cells.
- A DNA repair gene failure can both cause cancer and create a therapeutic vulnerability through synthetic lethality.
- BRCA-driven homologous recombination deficiency is the prototypical example leveraged by PARP inhibitors.
- Resistance emerges through multiple mechanisms; monitoring and combination strategies are critical.
- Comprehensive genomic and functional testing improves patient selection and outcomes.
- Future therapies will blend targeted agents, immunotherapy, and adaptive treatment plans to extend durable control.
Conclusion: Turning Repair Failure into an Advantage
The story of a DNA repair gene gone rogue is a study in biological irony. What begins as a genetic misfortune that enables cancer also creates a point of fragility clinicians can exploit. The translation of that insight into effective therapies illustrates the power of precision biology: understand the tumor’s weaknesses, tailor the attack, and stay one step ahead of resistance. As tools for detection and intervention improve, the hope is that many more cancers will be tackled not by brute force but by exploiting the very mistakes that allowed them to arise.