Surprising Molecular Link Between Cancer and Alzheimer's
The headline is arresting: mechanisms once thought to lie at opposite ends of human biology—unchecked cell proliferation on the one hand, and progressive neuronal loss on the other—may in fact be two sides of the same molecular coin. Scientists have uncovered evidence that key pathways involved in cancer biology also play central roles in the cascade that leads to Alzheimer's disease. The finding reframes how we think about aging, disease risk, and therapeutic strategy, and it carries implications for millions living with or at risk for either condition.
Why this matters
At first glance, cancer and Alzheimer's disease could not be more different. Cancer is defined by cells that refuse to die and divide without restraint. Alzheimer's is defined by progressive loss of neurons and cognitive decline. Yet the unexpected intersection of these diseases suggests that interventions aimed at one might influence the other, for better or worse. That possibility matters for clinicians, researchers, patients, and policymakers because it reshapes priorities for drug development, screening, and public health messaging.
What researchers actually found
Teams studying the molecular biology of neurodegeneration report that proteins and pathways central to cancer—cell-cycle regulators, tumor suppressors, and senescence-associated signals—also influence neuronal survival, protein aggregation, and brain inflammation. Laboratory models show that manipulations of molecules like p53, PIN1, and pathways that control senescence, DNA repair, and immune surveillance can simultaneously affect tumor formation and neural resilience. Those findings are not a single headline-making experiment but a tapestry of results from genetics, animal models, and tissue studies that converge on shared biology.
"A surprising overlap in molecular logic suggests that cancer and Alzheimer's are linked by how cells respond to damage, stress, and age."
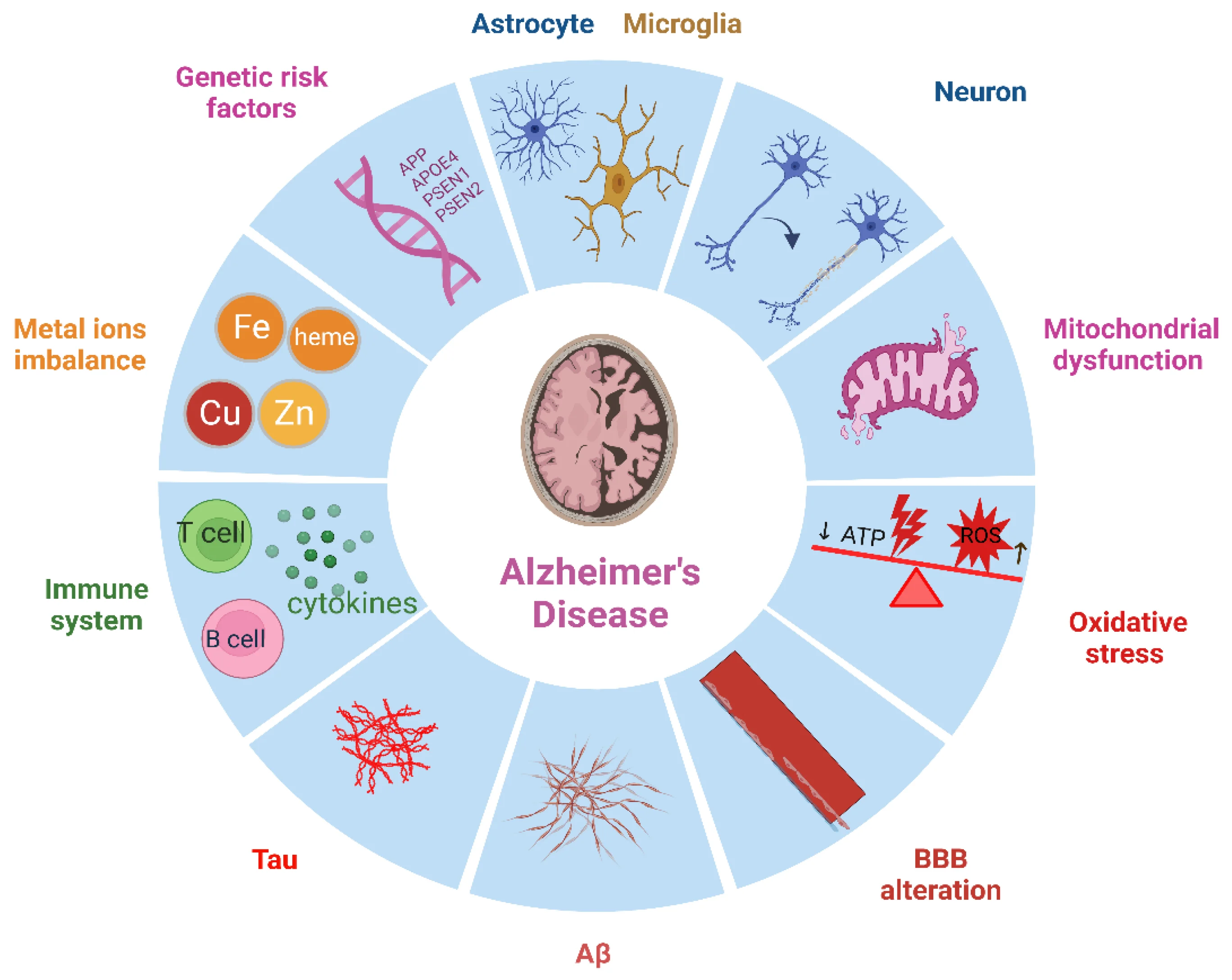
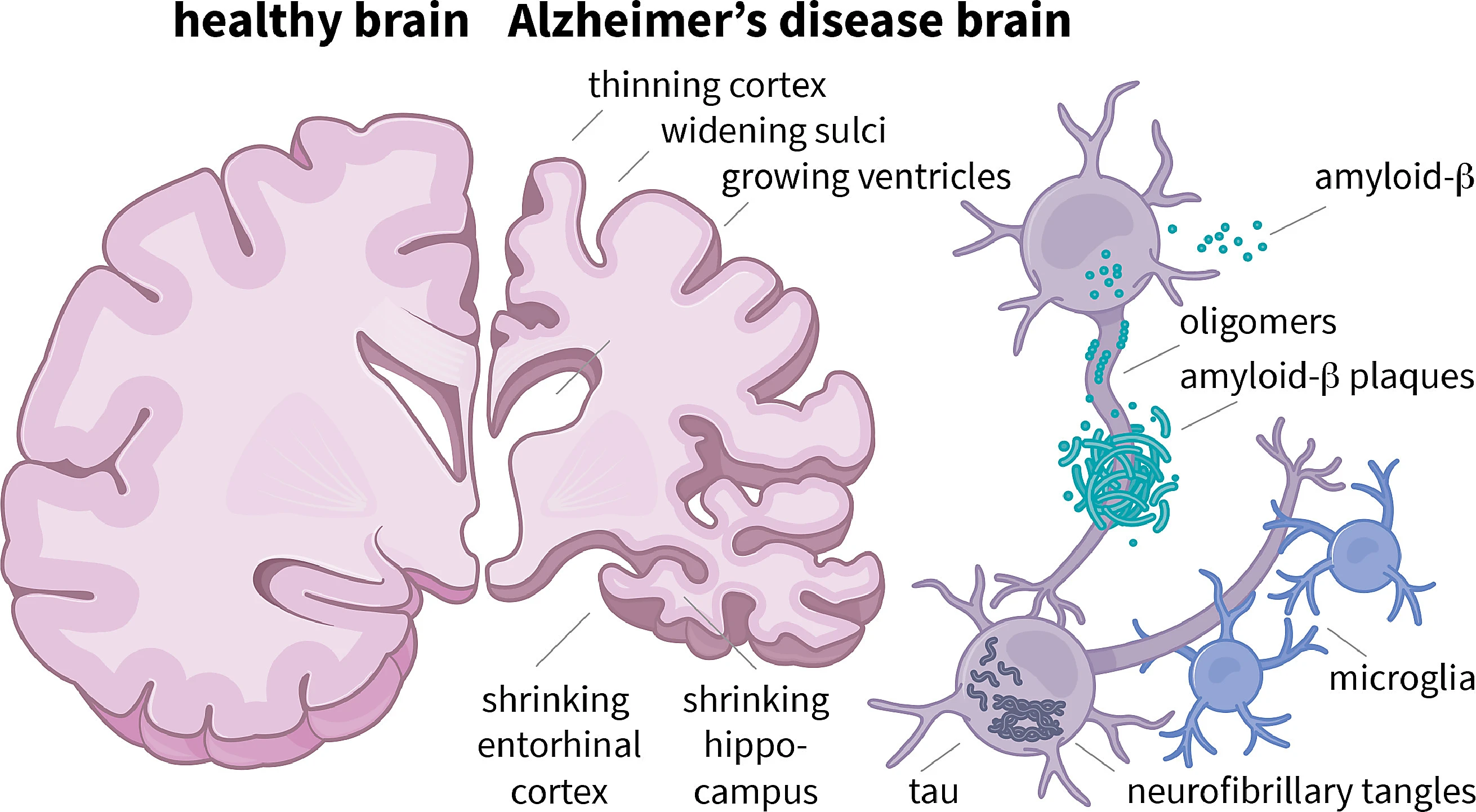
Understanding Alzheimer's: a quick primer
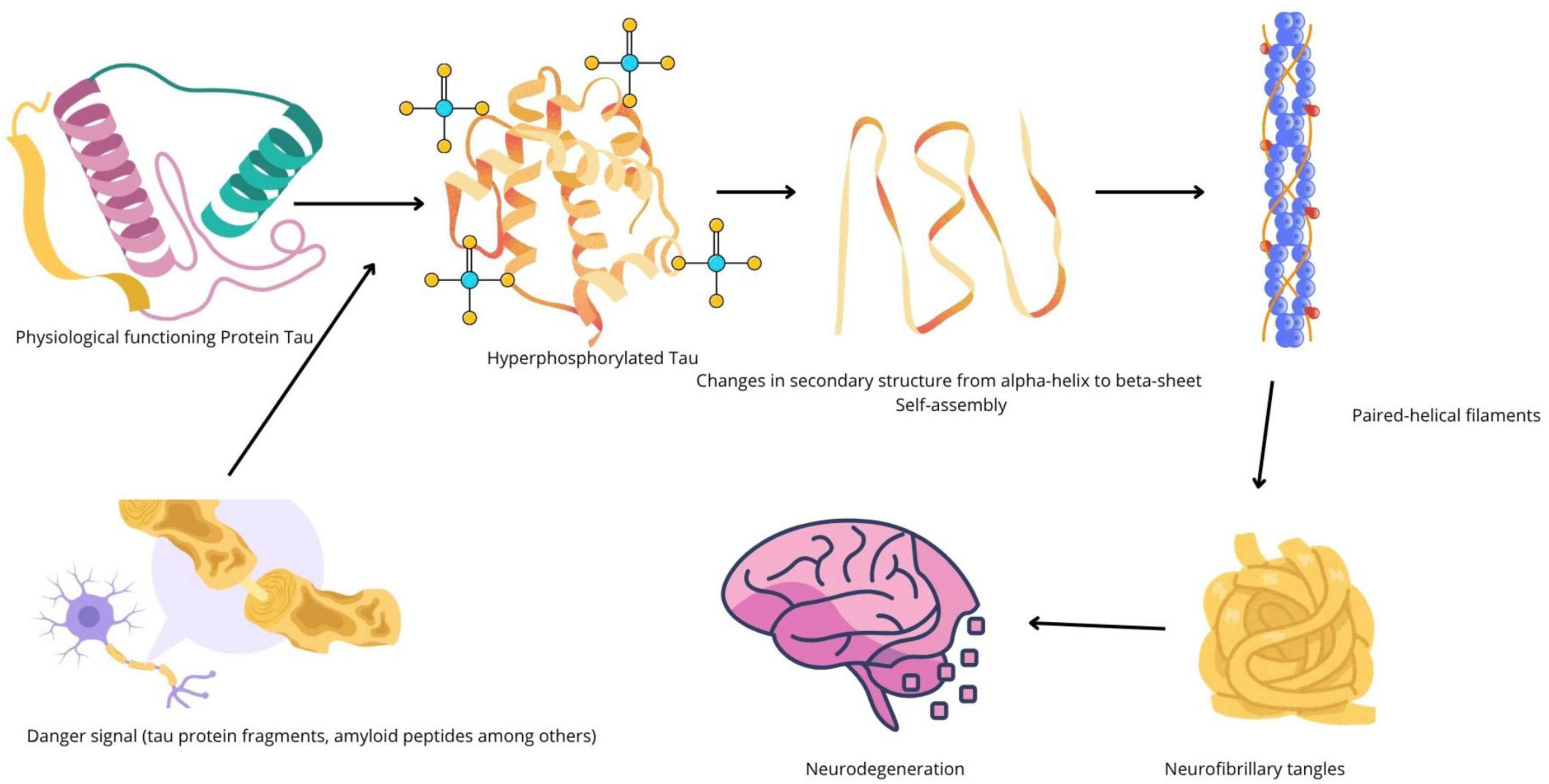
Alzheimer's disease is a progressive neurodegenerative disorder typically characterized by memory loss, impaired judgment, language difficulties, and eventual loss of independence. Microscopically, two features dominate: extracellular plaques composed of amyloid-beta peptides and intracellular tangles made of hyperphosphorylated tau protein. Those aggregates disrupt synaptic function, alter neuronal metabolism, and trigger inflammatory responses mediated by microglia and astrocytes. Over time, brain circuits collapse and cognitive symptoms worsen.
Alzheimer's amyloid-beta plaques
Tau protein neurofibrillary tangles
Understanding cancer: the other side of the coin
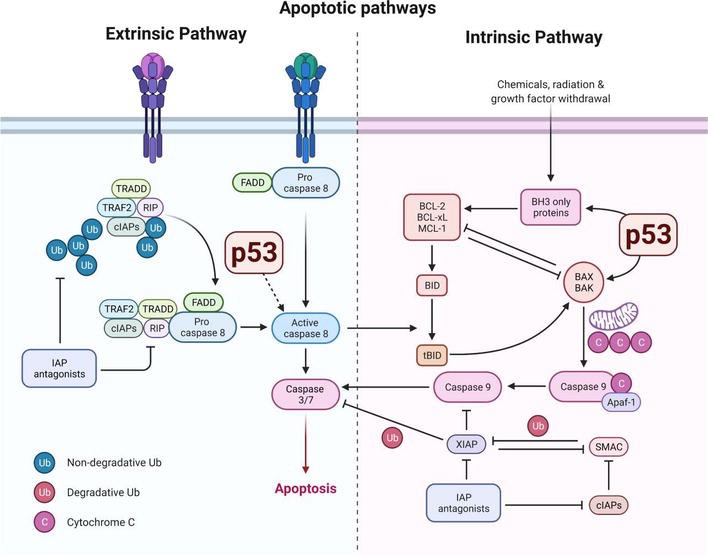
Cancer arises when cells acquire mutations and epigenetic changes that disable normal controls over growth, division, and death. Key processes include evasion of apoptosis (programmed cell death), dysregulated cell-cycle progression, genomic instability, and altered interactions with the immune system. Tumor suppressors such as p53 and regulators of the cell cycle stand guard against malignant transformation; when they falter, the risk of cancer increases.
p53 tumor suppressor protein
Where the two intersect: shared molecules and pathways
Scientists now point to several overlapping mechanisms that can plausibly link cancer and Alzheimer's. The clearest themes are cell-cycle control, protein quality and turnover, DNA damage responses, cellular senescence, and immune signaling.
Cell-cycle proteins and neuronal vulnerability
Neurons are largely post-mitotic: they do not normally divide. Yet many studies show that neurons under stress can aberrantly re-enter parts of the cell cycle, a process that often precedes cell death. Proteins that regulate the cell cycle in dividing cells—cyclin-dependent kinases, CDK inhibitors, and associated checkpoints—therefore have outsized effects on neuronal survival when they are misregulated. The same proteins are well established in cancer biology as gates for proliferation, so their dysregulation is a molecular bridge between uncontrolled division and neuronal loss.
Tumor suppressors and neurodegeneration: p53 as a double agent
p53 is one of the best-known tumor suppressors: it senses DNA damage and either halts the cell cycle to permit repair or triggers apoptosis if damage is irreparable. In the brain, p53 activity appears more nuanced. Overactivation of p53 in neurons can promote death and contribute to neurodegeneration. Conversely, loss of p53 function in peripheral tissues removes a check against cancer. Thus, p53 illustrates a tension: robust activity prevents cancer but may increase vulnerability to neurodegeneration, while weakened p53 reduces neuronal stress responses and increases cancer risk.
Protein homeostasis: amyloid, tau, and proteostasis networks
Protein folding, clearance, and the systems that manage misfolded proteins—collectively called proteostasis—are central to both diseases. In Alzheimer's, failures in proteostasis lead to amyloid-beta and tau accumulation. In cancer, proteostasis networks are co-opted to allow malignant cells to tolerate abnormal protein loads and the stress of rapid division. Elements like the ubiquitin-proteasome system, autophagy pathways, and chaperone proteins are therefore implicated in both pathologies, which suggests that drugs modulating these systems could have opposing effects depending on context.
Cellular senescence and the aging brain
Cellular senescence is a state in which cells stop dividing but secrete a cocktail of inflammatory and tissue-altering factors called the senescence-associated secretory phenotype (SASP). Senescent cells accumulate with age in many tissues and can promote chronic inflammation. In the brain, senescent glial cells appear to contribute to neuroinflammation and synaptic dysfunction, which can accelerate cognitive decline. Meanwhile, senescence suppresses cancer by arresting problematic cells. This creates another biological tradeoff: senescence is tumor-protective but may fuel degenerative processes in the aging brain.
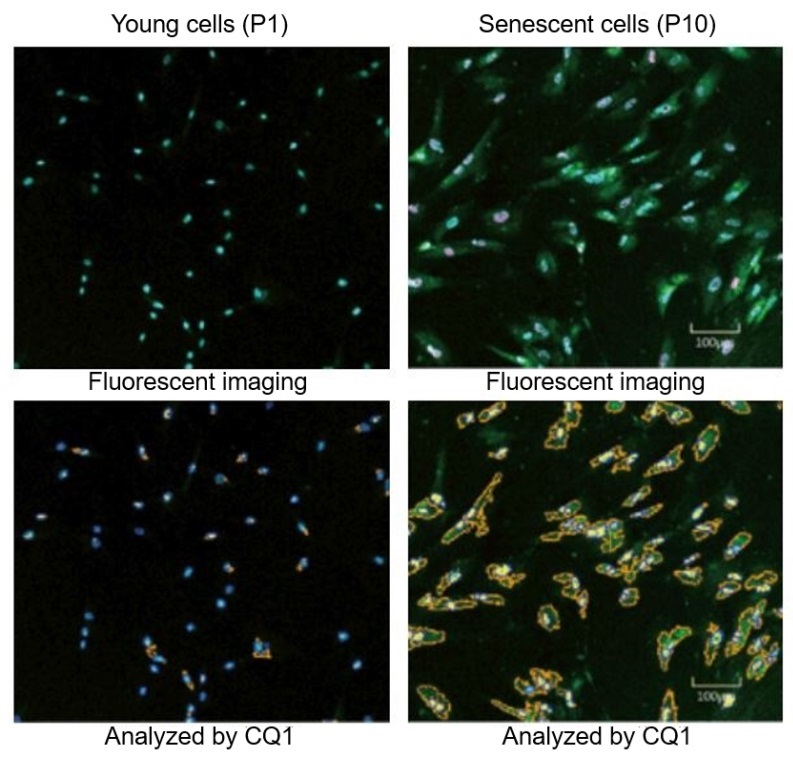
Cellular senescence microscopy
Immune surveillance and neuroinflammation
The immune system surveils tissues for mutated cells and clears emerging tumors. In the brain, microglia perform immune-like roles but also modulate synaptic pruning and respond to protein aggregates. Chronic activation of immune cells can damage neurons and promote Alzheimer's pathology, yet effective immune surveillance is essential to prevent cancer. Recent work suggests that shifts in immune function with age can simultaneously decrease anti-tumor responses and increase neuroinflammation, further tying the two conditions together.
Microglia neuroinflammation brain
The epidemiological paradox: inverse associations
Large observational studies have documented an intriguing epidemiological pattern: people with a history of certain cancers appear to have a lower than expected risk of Alzheimer's disease, and some patients with Alzheimer's show decreased cancer incidence. This inverse association is not universal and varies by cancer type and study design, but it has been reported across multiple cohorts. The paradox may reflect competing molecular mechanisms, survival bias, or differences in surveillance and reporting. Parsing these possibilities is a key challenge for researchers.
How to interpret the paradox
Several hypotheses could reconcile the inverse epidemiology. One is biological tradeoffs: pathways that protect against cancer (such as heightened senescence or active tumor suppressors) might increase Alzheimer's risk. Another is detection bias: cancer patients may die earlier and not live long enough to develop Alzheimer's, or follow-up and diagnostic patterns differ between patient groups. Finally, some cancer treatments may alter long-term cognitive trajectories either positively by clearing senescent cells or negatively through neurotoxic effects.
"The paradox forces us to ask whether preventing one disease might unintentionally influence the other."
Clinical implications: what this means for treatment and research
The intersection of cancer and Alzheimer's biology opens both opportunity and caution. On one hand, drugs developed for oncology may be repurposed to modify neurodegenerative processes. On the other hand, interventions designed to prevent or treat Alzheimer's might influence cancer risk. Thoughtful translational work will be required to evaluate both benefits and unintended harms.
Drug repurposing and therapy design
Some oncology agents target cell-cycle machinery, proteostasis, or senescence pathways—making them logical candidates for testing in neurodegeneration models. Senolytic drugs, which selectively clear senescent cells, are particularly promising because they could reduce neuroinflammation. However, senolytics could theoretically remove a cancer-protective brake in some tissues, so safety studies must be rigorous and long term.
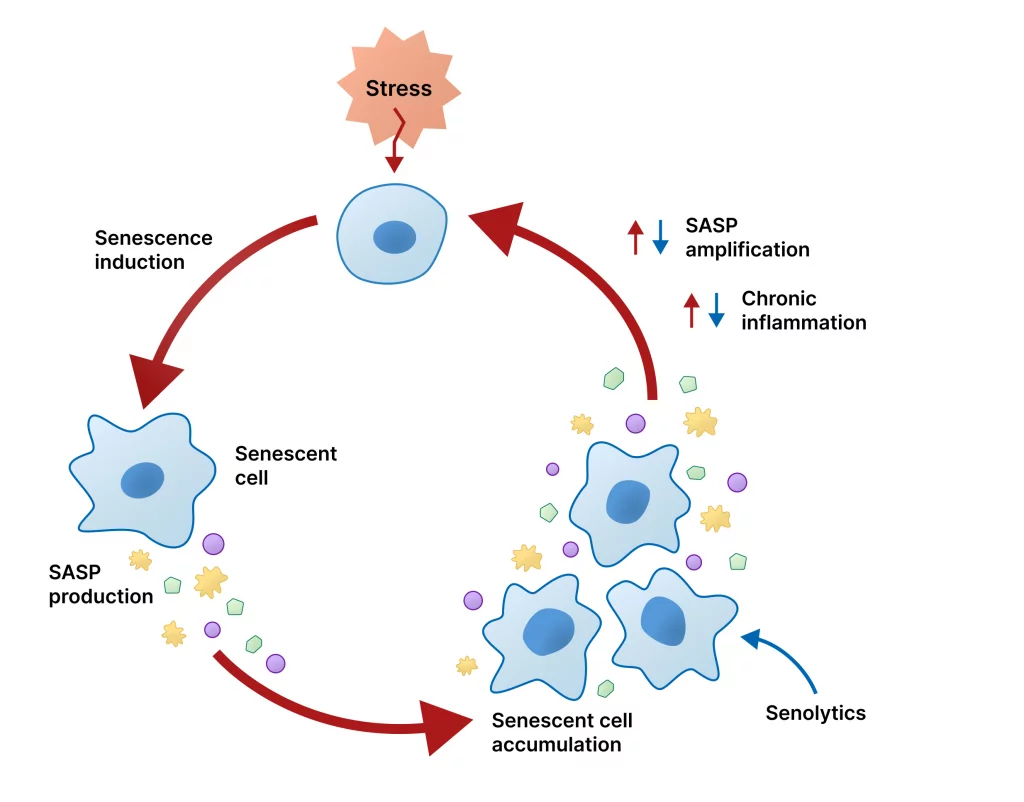
Senolytic drugs mechanism
Biomarker opportunities
Shared molecular players create opportunities to develop biomarkers that predict risk across diseases. Blood-based assays for protein aggregates, markers of DNA damage, or signatures of immune activation could inform both oncology and neurology clinics. Unified biomarker strategies would also make clinical trials more efficient by identifying populations most likely to benefit.
Clinical trial design: balancing competing risks
Future trials will need to stratify participants by cancer history, genetic risk factors, and biomarkers of senescence and immune function. Adaptive designs and careful long-term follow-up will be essential to detect both therapeutic benefits for cognition and any shifts in cancer risk. Interdisciplinary consortia that include oncologists, neurologists, geriatricians, and immunologists will be critical.
Practical advice for patients and clinicians
For now, the discovery is provocative but not prescriptive. It does not mean that people should change treatments without consulting clinicians. Nonetheless, it highlights the importance of integrated care for older adults: a comprehensive view that considers cognitive health, cancer screening, and the broader context of biological aging.
What patients should ask
Patients and caregivers can ask their doctors about how a history of cancer might influence cognitive health and vice versa. Key questions include whether certain medications could affect both conditions, whether additional monitoring is advisable, and whether participation in clinical research is a suitable option. Personalized risk assessment—incorporating genetics, family history, and biomarkers—remains the best path forward.
Ethical, social, and research challenges
The convergence of cancer and Alzheimer's biology raises difficult questions. If a drug reduces Alzheimer's risk but increases cancer risk, how should clinicians weigh those outcomes with patients? How should public health messaging balance prevention strategies? Research must approach these questions with transparency, thorough risk–benefit analysis, and inclusion of diverse populations that historically have been underrepresented in both oncology and neurology trials.
Equity and access
Biomarker-driven strategies and new therapeutics risk widening disparities unless access is built into design and policy. Ensuring that advances benefit all communities will require intentional outreach, affordable testing, and equitable trial enrollment.
Where the field goes next
Researchers will pursue several parallel paths: deeper mechanistic studies to map overlapping pathways, translational work to test candidate drugs in relevant models, and epidemiological research that teases apart bias from biology. Large, longitudinal cohorts with linked clinical, genetic, and biomarker data are particularly valuable. Cross-disciplinary training programs will help bridge the language and practice gaps between oncology and neurodegeneration research.
A roadmap for researchers
Immediate priorities include: (1) validating molecular targets across model systems, (2) developing safe dosing strategies for agents that affect cell-cycle and senescence pathways, (3) creating sensitive, noninvasive biomarkers to monitor both neurodegeneration and cancer risk, and (4) designing trials that measure long-term outcomes beyond the primary endpoint.
Conclusion: a new way of thinking
The discovery of overlapping biology between cancer and Alzheimer's reframes two major public-health challenges as interconnected outcomes of aging and cellular stress. It does not collapse the distinction between the diseases, but it does demand a more integrated view of biology, prevention, and treatment. For patients, the message is one of cautious optimism: understanding shared pathways could accelerate discovery and lead to interventions that preserve both lifespan and brain health. For researchers and clinicians, the message is to collaborate across disciplines, think longitudinally, and design studies that explicitly consider the tradeoffs inherent in aging biology.
- Core molecular pathways—cell-cycle control, proteostasis, senescence, and immune signaling—are implicated in both cancer and Alzheimer's disease.
- Observational studies show complex, sometimes inverse epidemiological relationships between cancer and Alzheimer's, which may reflect biological tradeoffs or bias.
- Translational opportunities exist for drug repurposing and biomarker development, but safety and long-term effects must be rigorously assessed.
- Integrated, cross-disciplinary research and equitable clinical strategies are essential to translate these findings into better outcomes.
This article synthesizes current concepts linking cancer biology and neurodegeneration to inform clinicians, researchers, and the public. Consult medical professionals before making treatment decisions.