New Clues: How Alzheimer's Destroys Neurons
The headline feels almost too good to be true: after decades of dead ends and partial answers, scientists may finally be closing in on the precise sequence of events that turns a living neuron into a corpse in Alzheimer's disease. What was once a blurred field of competing theories—amyloid plaques, tangles of tau protein, chronic inflammation, metabolic failure—now looks more like an interconnected cascade. The breakthrough is not a single magic bullet, but a clearer map of the chain reaction that links protein pathology to immune activation and, ultimately, a form of regulated cell death. That map changes how we evaluate therapies, how we design clinical trials, and how families understand what is happening inside a loved one's brain.
Alzheimer's brain neurons
What researchers have found
A converging picture
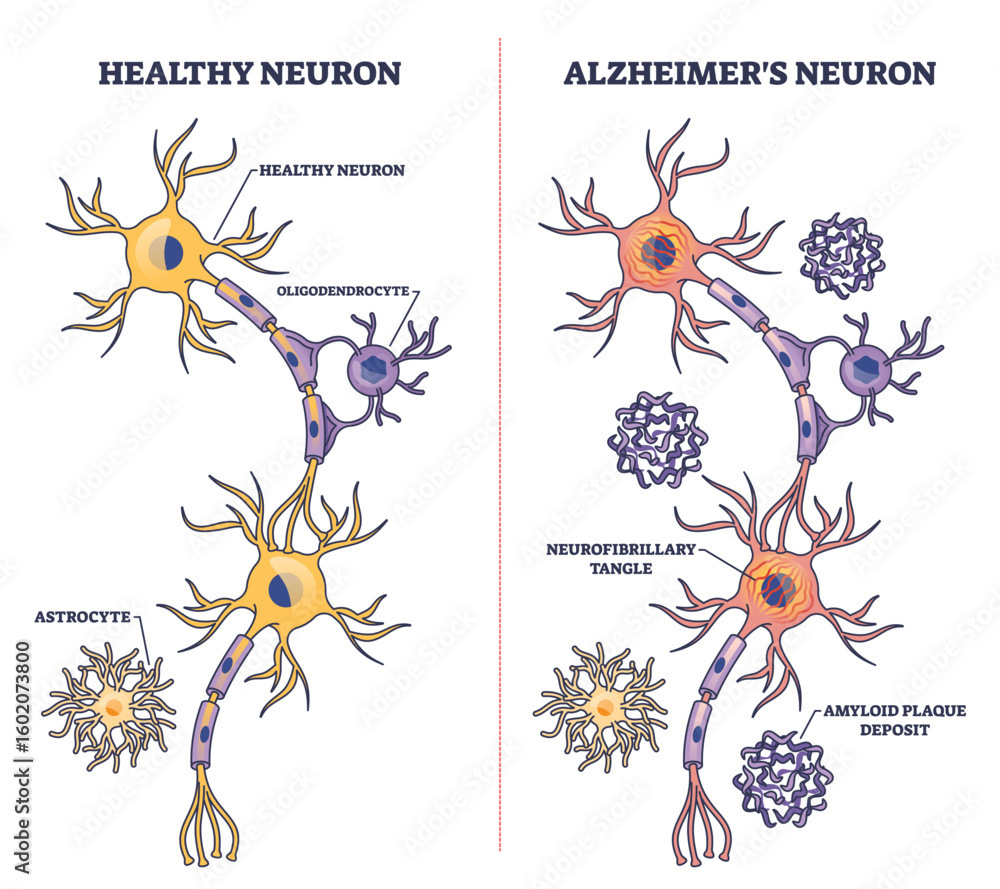
For years Alzheimer's research has been split into camps. One camp emphasized extracellular amyloid-beta as the initiating insult; another focused on intracellular tau tangles as the proximate toxin. Increasingly, the field recognizes both as parts of a sequence: misfolded proteins disturb normal neuronal function, and that disturbance recruits the brain's innate immune cells—microglia—which in turn amplify damage through inflammatory signaling and synapse removal. The latest insights add a critical final step to this progression: neurons, after suffering chronic stress and immune-mediated assault, activate internal death programs that are distinct from the random necrosis once imagined. In short, Alzheimer's kills neurons by triggering regulated, self-directed death pathways that are facilitated and sometimes initiated by immune activity and protein pathology.
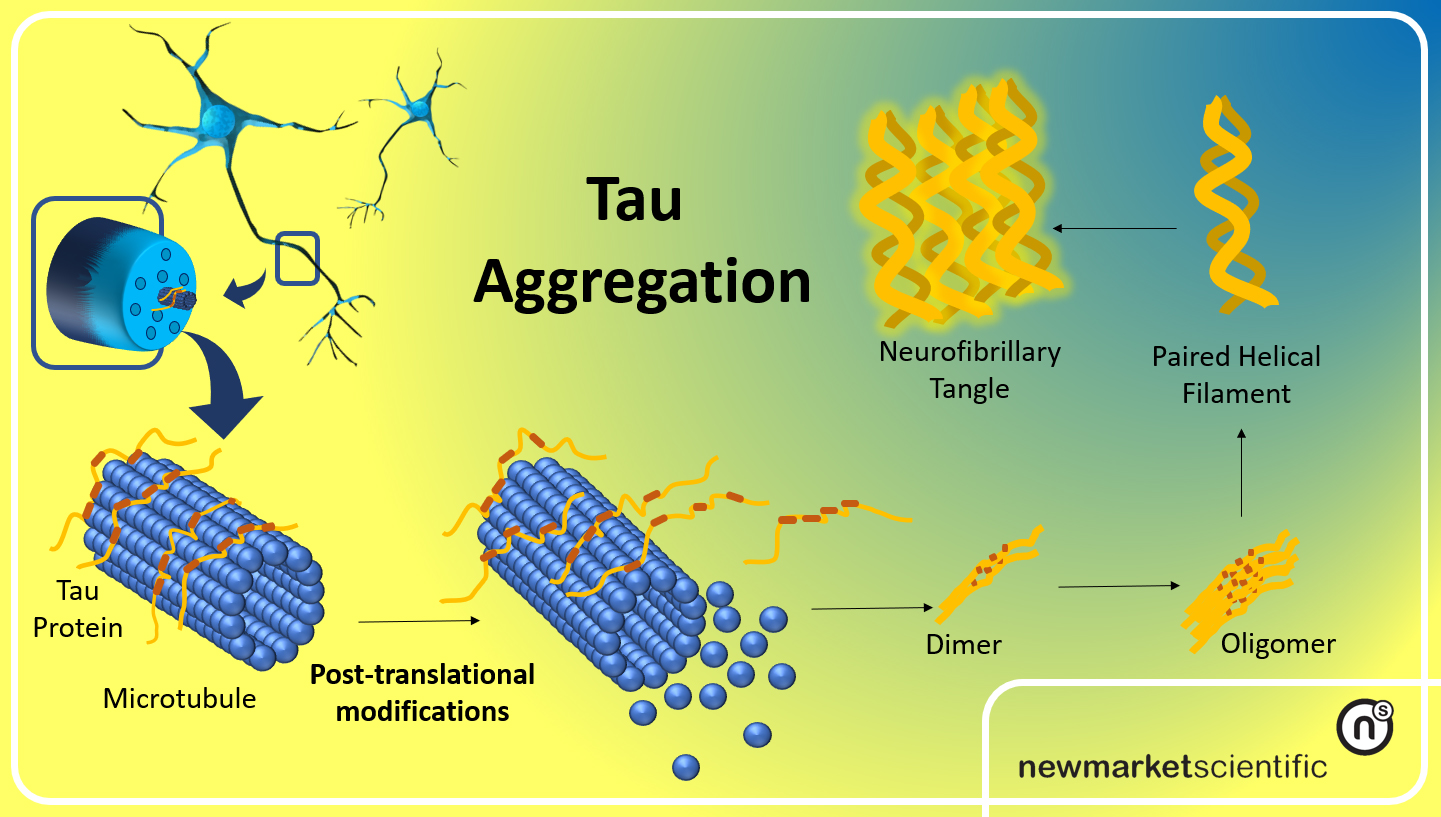
tau protein amyloid aggregation
The players and the cascade
At the center of this cascade are a few recurring elements: misfolded tau and amyloid proteins, reactive microglia, complement proteins that tag synapses, and molecular machines inside neurons that execute distinct death programs. Misfolded tau impairs axonal transport and synaptic signaling, making neurons dysfunctional. Microglia sense these dysfunctions and move in to prune damaged synapses, a process normally useful during development but destructive when chronic. Complement proteins such as C1q and C3 can mark synapses for removal. Prolonged pruning and sustained inflammatory signaling stress neuronal metabolism and mitochondria, pushing cells toward regulated death mechanisms—examples include apoptosis-like cascades and, importantly, necroptosis, a programmed, inflammatory form of cell death.
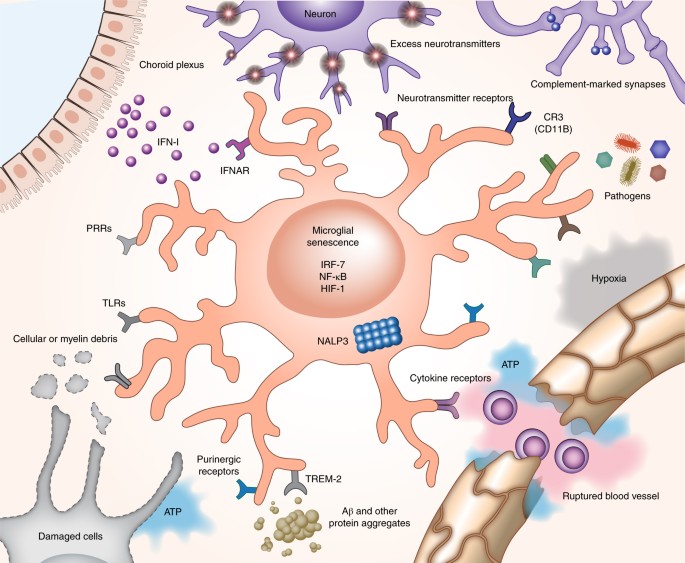
microglia immune brain cells
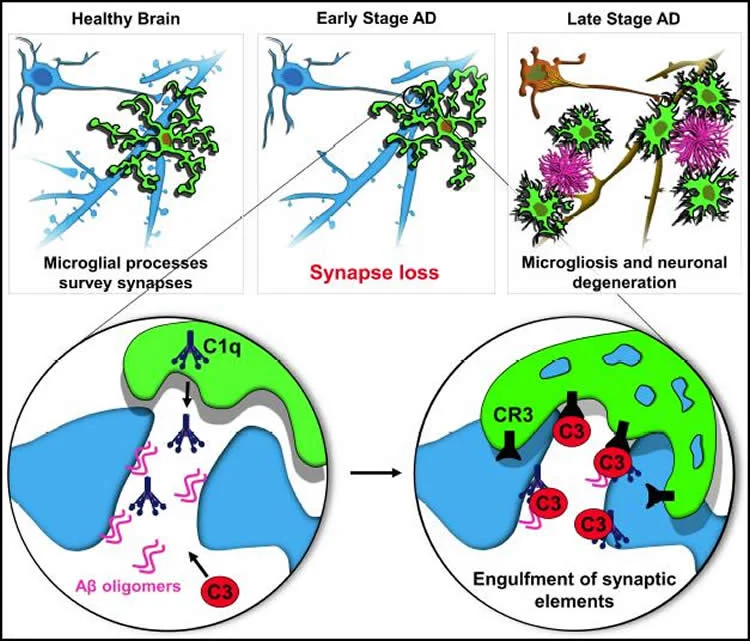
synaptic loss Alzheimer's disease
When the brain's clean-up crew becomes a demolition team, neurons don't just fail — they are actively dismantled.
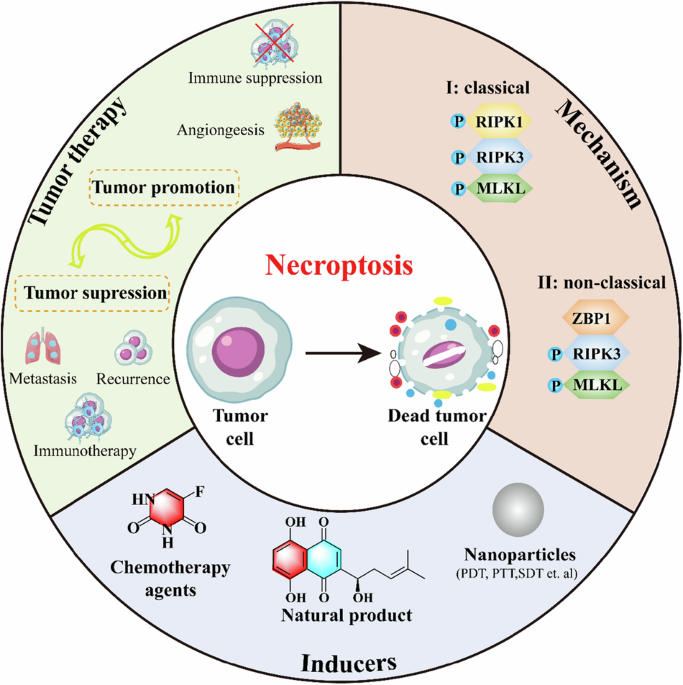
necroptosis programmed cell death
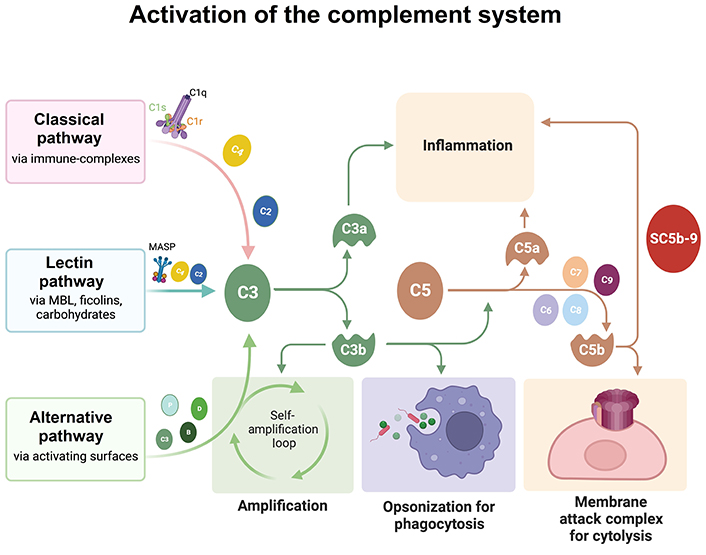
complement system C1q proteins
How scientists reached this view
Techniques that made the invisible visible
The modern picture has emerged because several technologies matured at once. Single-cell and single-nucleus RNA sequencing allowed researchers to read the transcriptional state of individual neurons and glia, revealing signatures of stress, immune activation, and death pathways in specific cell types. Spatial transcriptomics added the crucial context of where these states occur in the tissue. Advanced proteomics exposed which proteins are chemically modified or aggregated. Induced pluripotent stem cell (iPSC) models let investigators recreate human neurons and glia in a dish, testing how human cells respond to tau or amyloid. High-resolution imaging captured synapse loss and microglial behavior in real time in animal models. Taken together, these methods make it possible to move from correlation to mechanism: seeing not only that tau and microglial markers coexist, but that microglial activation precedes or accompanies neuronal death signatures in the same microenvironments.
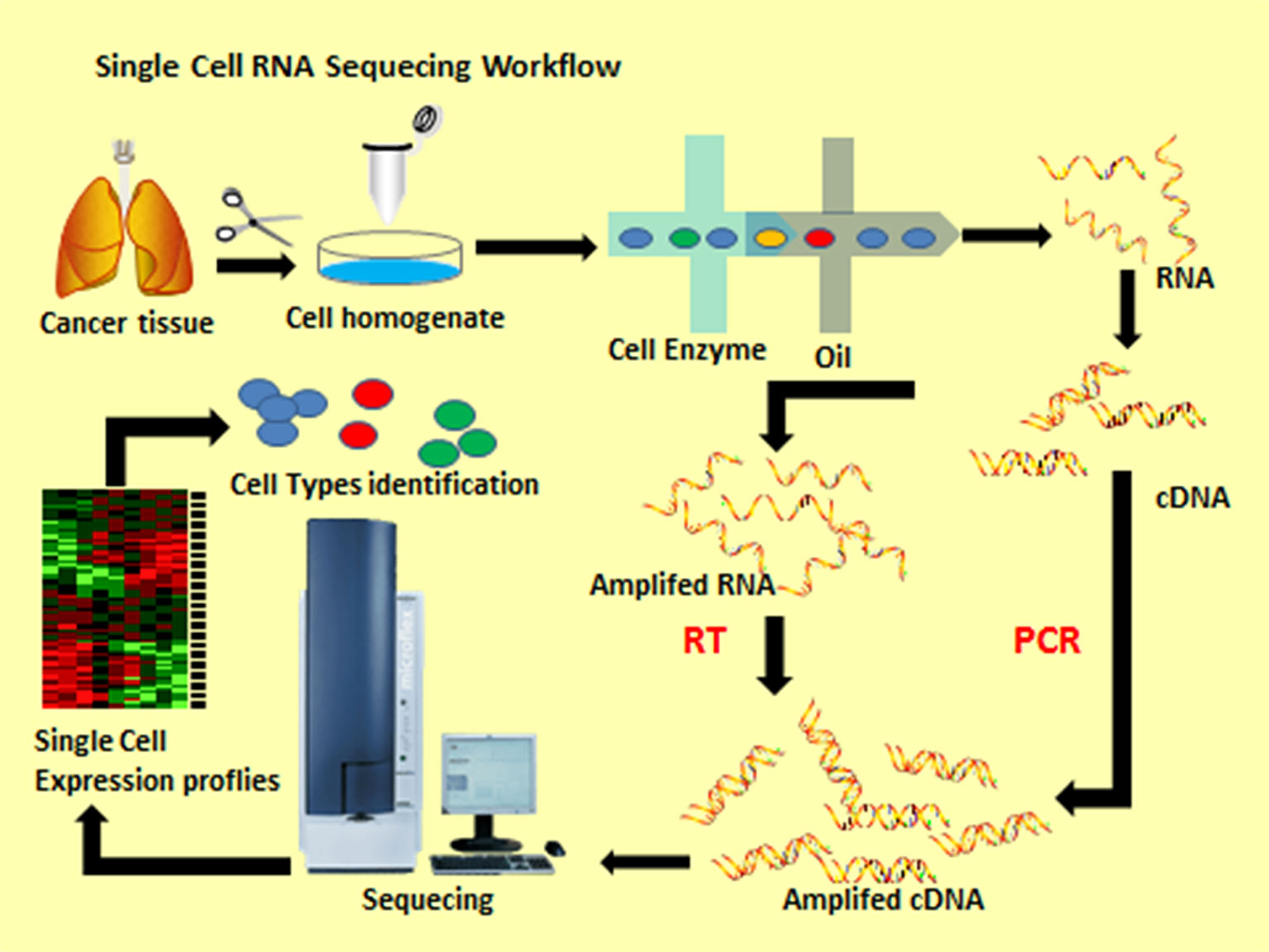
single-cell RNA sequencing technology
Experimental evidence: models and patient tissue
Robust conclusions come from converging lines of evidence. In experimental animals and human tissue samples, researchers have observed the same basic sequence: protein pathology, microglial activation, complement tagging, synaptic loss, and then neuronal death signatures. Manipulating one node in the chain—reducing complement activity, dampening microglial inflammation, or blocking key death-execution proteins—often attenuates subsequent steps in the cascade. Those experiments don't yet prove every detail, and species differences remain, but they provide a consistent, testable framework.
Why this changes the therapeutic landscape
From single target to sequence interruption
Therapeutic thinking shifts when disease is understood as a cascade. If neuron death results from a multi-step process, then therapies can aim to interrupt the sequence at several points: reduce protein misfolding, block harmful microglial activation, prevent complement-mediated pruning, or inhibit the final executioners of cell death. Each intervention has pros and cons. Targeting protein aggregation may be most effective early; modulating microglia might protect synapses even after pathology appears; inhibiting necroptosis or related death pathways could preserve neurons in mid-stage disease. The key is matching intervention to disease stage, and perhaps combining approaches.
- Multiple drug targets increase chances of effective therapy.
- Stage-specific interventions allow personalized treatment plans.
- Increased complexity makes trials harder to design.
- Off-target immune effects risk impairing beneficial brain functions.
Examples of actionable targets
Several classes of interventions now look particularly compelling. Small molecules that inhibit kinases involved in necroptosis or other death pathways could blunt neuronal loss. Antibodies or small biologics that neutralize damaging microglial signals or block complement tagging may protect synapses. Approaches to stabilize tau folding, enhance protein clearance by the lysosome-autophagy system, or restore mitochondrial health are also complementary. Importantly, some of these approaches are already in early clinical testing; others remain preclinical but attractive.
Practical challenges and uncertainties
Heterogeneity and timing
No single model of Alzheimer's fits every patient. The disease varies by genetic background, coexisting vascular disease, metabolic state, and other factors like age and lifestyle. That heterogeneity means an intervention that helps one subgroup may fail in another. Timing is equally critical: an anti-pruning therapy might help preserve synapses in early disease, but could be less effective if neurons have already passed the point of no return. Distinguishing these stages in living patients requires better biomarkers.
Risks of modulating the immune system
The brain's immune system performs essential housekeeping, so blunting microglia or complement indiscriminately could interfere with normal function and increase infection risk. Any therapy that targets immune pathways must be designed to restore balance rather than eliminate function. Achieving that balance will require precise dosing, careful patient selection, and thoughtful monitoring.
What this means for people living with Alzheimer's
Realistic hope, realistic timelines
These mechanistic advances are promising, but they do not translate into an immediate cure. Drug development is slow and expensive; even with a validated target, successful drugs often take years in clinical trials. Still, a clearer map of how neurons die allows for smarter trials: better selection of patients, more sensitive biomarkers to detect biological impact, and combination therapies that address different cascade steps. For patients and families, the near-term benefit may be trials that are more informative and a sense that science is finally aligning around actionable mechanisms.
What to watch for
Over the coming years, pay attention to trials that specifically target microglial activation, complement pathways, or necroptosis-related proteins, and to studies that pair these interventions with biomarkers showing reduced synapse loss or neuronal stress. Biomarkers—blood-based and imaging—will be crucial in determining whether a therapy slows the cascade in living patients.
Next steps for research
Refining causality and translation
Important work remains. Scientists must continue to test causality: if you block a proposed step, does neuronal survival improve and do cognitive outcomes follow? Translational research must bridge the gap between animal models and human brains—especially older brains with multiple pathologies. Improved biomarkers that reflect specific cascade steps rather than broad damage will permit adaptive trials that learn quickly whether a mechanism is being hit.
Broadening the toolbox
The toolkit for intervening will expand beyond classic small molecules and antibodies. Gene-silencing approaches, precision biologics that tweak microglial signaling without shutting it down, and repurposed drugs that stabilize mitochondria or lysosomes may all play a role. Successful strategies will likely combine approaches to reduce ongoing damage while bolstering neuronal resilience.
Conclusion
The emerging view—that Alzheimer's disease kills neurons through a linked sequence of protein misfolding, immune-mediated synaptic destruction, and regulated neuronal death—brings both clarity and complexity. It reframes the disease from a single villain to a process, and that reframing is liberating: it multiplies the therapeutic opportunities and forces a more nuanced approach to trials and treatment. For patients and families the message is one of cautious optimism. Science has not yet delivered a cure, but it has sketched a much more detailed battlefield plan. The next decade will be about turning that plan into effective, stage-appropriate therapies and reliable biomarkers that let doctors intervene before the cascade becomes irreversible.
Detailed maps of the death cascade give us better targets—if we act at the right time.
- Alzheimer's appears to involve a cascade linking misfolded proteins, immune activation by microglia, complement-mediated synapse loss, and regulated neuronal death.
- Understanding the sequence creates multiple intervention points—early anti-aggregation therapy, mid-stage immune modulation, and late-stage cell-death inhibition.
- Translating these insights into treatments requires precise biomarkers, careful trial design, and attention to patient heterogeneity.